
At Streamlit, we always look for ways to improve your experience and make app development simple. Custom components are essential to this, as they provide a way to contribute to and extend the Streamlit ecosystem. We wanted to create something useful for the community and show how easy it is to write a custom component.
Today, we are excited to introduce a new custom component for Streamlit...
...the molecule editor! 🧪
The ability to draw chemical compounds is critical for most drug discovery, drug design, and cheminformatics apps. You can now easily integrate a molecule editor into your Streamlit applications with just a few lines of code.
How to use Streamlit molecule editor
First, install Streamlit and the component:
pip install streamlit
pip install streamlit-ketcher
Then, import it into your Streamlit application, and you're good to go:
import streamlit as st
from streamlit_ketcher import st_ketcher
smiles = st_ketcher()
This code snippet creates a full-featured chemical molecule editor within your Streamlit app. The variable smiles contains the Simplified Molecular Input Line Entry System (SMILES) representation of the molecule, which you can use for further processing, analysis, or visualization in your app.
Our component is based on the Ketcher library from Epam. Several other editors offer similar functionality, including MarvinJS, JSME, and ChemWriter. However, we chose Ketcher for the following reasons:
- It's released under the Apache 2.0 license, compatible with the Streamlit library.
- It's React-friendly, as it has a corresponding React component.
- It has pretty good documentation.
- It has a modern look and feel and is feature-rich.
- Its most basic version doesn't rely on an external server.
So, it was the perfect fit for our needs. 🙂
Features
The molecule editor component offers several useful features, including:
- Intuitive drawing tools: Easily create and modify molecules using a simple point-and-click interface, with support for adding and deleting atoms, bonds, and functional groups.
- Automatic SMILES and Molfile generation: Instantly convert the molecule into its SMILES representation to easily integrate with other cheminformatics tools. Molfiles are also supported.
- Copy/paste support.
- Open-source and community-driven: The editor is free and open-source, allowing you to contribute and help improve the component.
Use cases
The editor is an excellent choice for a variety of cheminformatics and chemistry-related applications, including:
- Drug design and discovery
- Molecular modeling and visualization
- Management of chemical databases
- Education and training in chemoinformatics
- Prediction of chemical properties
Demo apps
We've prepared two demo applications.
Demo app 1 by Kamil Breguła
This app provides an overview of the basic usage and configuration of the component:

Demo app 2
This app explores some of a particular application's most popular use cases. It integrates with the open chemistry database ChEMBL using the chembl_webresource_client package (created a few years ago by the author of this doc). With this integration, molecule structures can be retrieved by name. The app can now create a gallery of the "most famous" chemical compounds at the top:

This is achieved with the following code:
famous_molecules = [
('☕', 'Caffeine'),
('🥱', 'Melatonin'),
('🚬', 'Nicotine'),
('🌨️', 'Cocaine'),
('💊', 'Aspirin'),
('🍄', 'Psilocybine'),
('💎', 'Lysergide')
]
for mol, column in zip(famous_molecules, st.columns(len(famous_molecules))):
with column:
emoji, name = mol
if st.button(f'{emoji} {name}'):
st.session_state.molfile, st.session_state.chembl_id = utils.name_to_molecule(name)
The name_to_molecule function is defined as follows:
from typing import Optional, Tuple
from chembl_webresource_client.new_client import new_client as ch
def name_to_molecule(name: str) -> Tuple[str, str]:
columns = ['molecule_chembl_id', 'molecule_structures']
ret = ch.molecule.filter(molecule_synonyms__molecule_synonym__iexact=name).only(columns)
best_match = ret[0]
return best_match["molecule_structures"]["molfile"], best_match["molecule_chembl_id"]
After you retrieve the molecule structure from ChEMBL and load it into the editor, you can modify it as needed. Once editing is complete, you can read the compound from the editor and run a similarity search to find the most similar compounds from ChEMBL. You can control the similarity threshold with a slider. The search results will be displayed in a table:
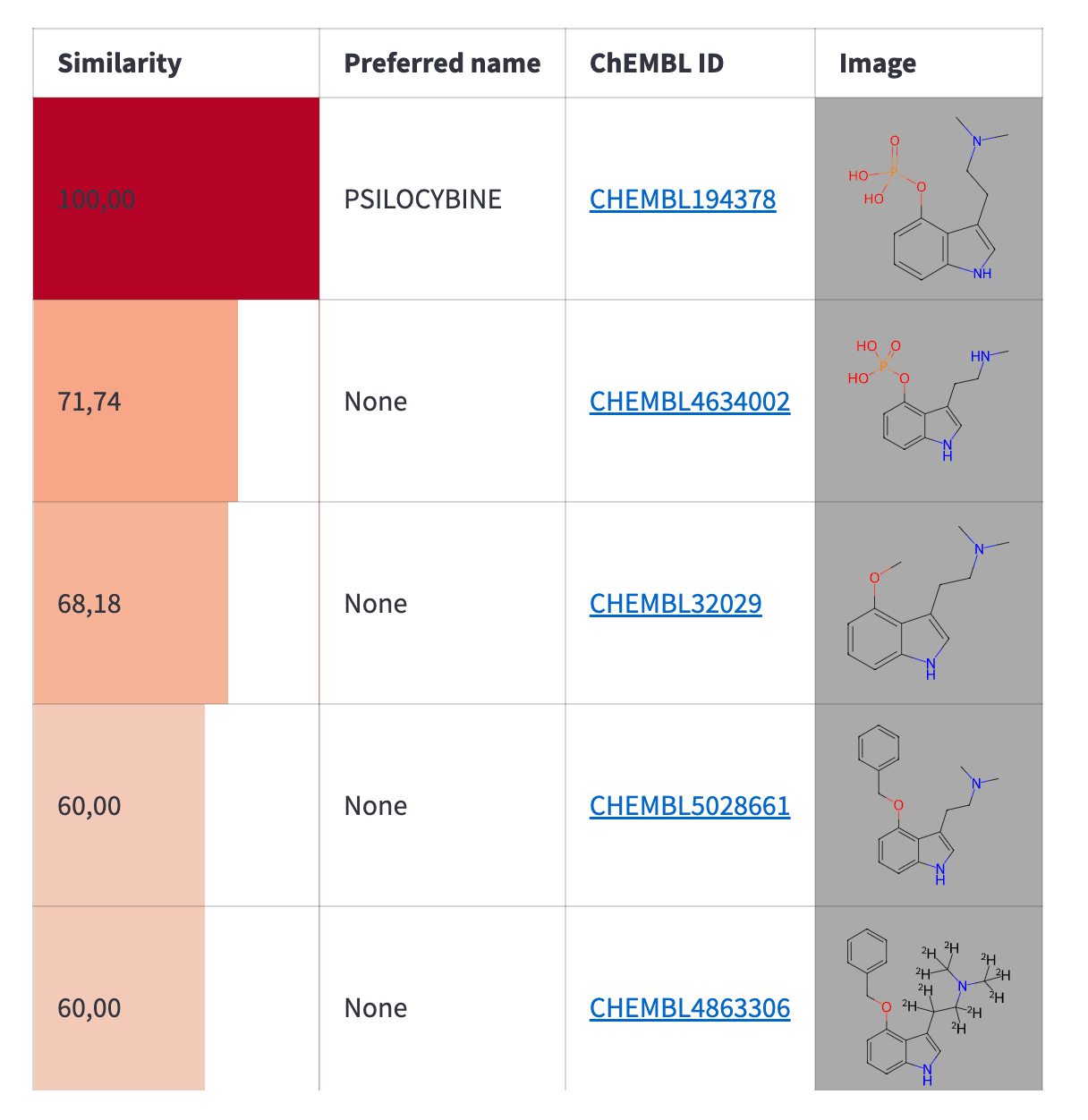
with editor_column:
smiles = st_ketcher(st.session_state.molfile)
similarity = st.slider("Similarity threshold:", min_value=60, max_value=100)
with st.expander("Raw data"):
st.markdown(f"```{smiles}```")
with results_column:
similar_molecules = utils.find_similar_molecules(smiles, similarity)
if not similar_molecules:
st.warning("No results found")
else:
table = utils.render_similarity_table(similar_molecules)
similar_smiles = utils.get_similar_smiles(similar_molecules)
st.markdown(f'<div style="overflow:scroll; height:600px; padding-left: 80px;">{table}</div>',
unsafe_allow_html=True)
You can perform the similarity search using the following function:
def find_similar_molecules(smiles: str, threshold: int):
columns = ['molecule_chembl_id', 'similarity', 'pref_name', 'molecule_structures']
try:
return ch.similarity.filter(smiles=smiles, similarity=threshold).only(columns)
except Exception as _:
return None
After retrieving compounds similar to the 'query compound' you drew in the editor, you can run a target prediction to determine which biological targets your compounds will bind to. To do this, use the onnxruntime library. It'll let you load a prediction model created by my EBI colleague, Eloy Felix (read more about model training and exporting to the ONNX format here).
All target prediction logic is implemented in the demo app's target_predictions.py module:

The result is a table of biological targets ranked by the probability of having an affinity for the predicted compounds. Note that some targets are repeated, as you get the results for each query compound separately. We omitted adding more information about the targets and filtering by organism or type since it's just a proof-of-concept app.
Are custom components easy to create?
The good news is that we created a fairly complex custom component in just a few days. This means that the current API is fully functional. But we've also identified some areas for improvement, including:
- Outdated component templates
- Undocumented React hook support
- Missing props in the theme interface definition
- Issues with the CSS that cause the component to render strangely without further customization
- Difficulty testing components
While we've found workarounds for most of these issues, we believe it's worth investing some time in the Custom Components ecosystem to make it more developer-friendly. What do you think? Have you ever created a custom component? Share your thoughts in the comments.
Wrapping up
We hope the molecule editor component is a valuable addition to your toolkit. We can't wait to see the amazing applications you create with it. Don't hesitate to contact us with questions or suggestions or to share your projects with the Streamlit community!
Happy coding! 🧑💻
References
- Streamlit Molecule Editor GitHub Repository: https://github.com/streamlit/streamlit-ketcher
- Ketcher Library by EPAM: https://lifescience.opensource.epam.com/ketcher
- ChEMBL database: https://www.ebi.ac.uk/chembl
- ChEMBL web resource client library: https://github.com/chembl/chembl_webresource_client
- ONNX runtime: https://onnxruntime.ai
- Simple demo code: https://ketcher-editor.streamlit.app/
- Advanced demo code: https://github.com/streamlit/mol-demo



Comments
Continue the conversation in our forums →